
Professor Stone has now retired from his university position
Although chemistry is concerned largely with reactions in which bonds are broken and reformed, the forces between unreacting molecules and fragments of molecules are often of great importance, for example in controlling the way in which protein molecules fold into their biologically-active structures. It has been understood for many years that the attractive forces between molecules are responsible for the very existence of condensed phases, but only very recently has it become possible to build sufficiently detailed models of these forces to explain the complex behaviour of real liquids. Even water, the most important liquid of all, is still very imperfectly understood. However our work has led to a description of the interactions between water molecules that was for some years the most accurate available, and which has led the way to more accurate intermolecular potentials for this and other systems.
Modern spectroscopic techniques provide detailed and accurate data that can be very helpful in refining both quantitative descriptions and qualitative understanding of intermolecular interactions. However, some applications require very accurate potentials, and experimental phenomena depend on the intermolecular potential in a complicated way, so it is difficult to extract accurate information about the potential from such data. For example, the prediction of crystal structures of organic molecules is important in the pharmaceutical industry, as there are often several possible crystal structures that differ in energy by a few kJ/mol or less, and it is necessary to find (and patent) the most stable one. Using modern computational techniques, however, it is becoming possible to calculate an accurate potential from first principles. The terms in the potential can be expressed in terms of molecular properties, in a simple form that allows macroscopic properties of liquids and crystals to be calculated. The main terms are electrostatic, repulsion, induction and dispersion. The electrostatic interaction depends on the charge distribution, and accurate descriptions, using ‘distributed multipoles’1 have been available for more than thirty years. The induction term requires polarizabilities, which describe the response of the molecular charge distribution to external fields, and although the theory of ‘distributed polarizabilities’ was developed 25 years ago, accurate methods for calculating them have only recently been developed. The same is true of the dispersion (van der Waals attraction) and repulsion terms, but it is now possible to calculate all of these terms and to construct accurate intermolecular potentials for molecules comprising 20 or so atoms.2
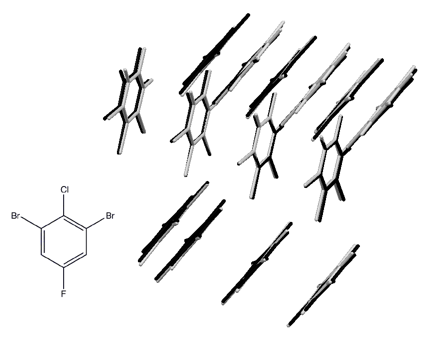
An early success of this approach was the prediction of the crystal structure of 1,3-dibromo 2-chloro 5-fluorobenzene from first principles (that is, without using any experimental data), as part of the 2007 blind challenge organized by the Cambridge Crystallographic Data Centre.3 The crystal structure was known, but was not revealed until after the predictions had been made, so this was a genuine prediction. The overlay shows the experimental structure (black) and the predicted structure (grey). The two structures are hard to separate as the overlay is nearly perfect.
More recently, we have developed an intermolecular potential for pyridine, a more difficult case because the dimer has several bound structures and the crystal has a number of polymorphs with several molecules, unrelated by symmetry, in the unit cell. Our first-principles potential4 was used successfully to predict a known but unpublished polymorph structure of crystalline pyridine5.
Selected Publications
- Anthony J. Stone, The Theory of Intermolecular Forces, 2nd edn 2013, (Clarendon Press, Oxford).
- Anthony J. Stone and Alston J. Misquitta, CamCASP: a program for calculating molecular properties and intermolecular interactions, http://www-stone.ch.cam.ac.uk/programs.html#CamCASP.
- Alston J. Misquitta, Gareth W.A. Welch, Anthony J. Stone and Sarah L. Price, A first principles prediction of the crystal structure of C6Br2ClFH2, Chem. Phys. Letters, 2008, 456, 105-109.
- Alston J. Misquitta and Anthony J. Stone, Ab initio atom--atom potentials using CamCASP: Theory and application to many-body models for the pyridine dimer, J. Chem. Theor. Comput., 2016, 12, 4184-4208.
- Alexander A. Aina and Alston J. Misquitta and Sarah L. Price, From dimers to the solid-state: Distributed intermolecular force-fields for pyridine, J. Chem. Pjys, 147, 161722.