06/02/2018
Andrea’s and Gerit’s paper has been published in the Proceedings of the National Academy of Sciences of the United States of America. The work had important contributions also from Jiří, a previous member of the ICE group, and from Dario and Alexandre, longstanding Angelos’ collaborators. In the paper we study molecular crystals with quantum Monte Carlo.
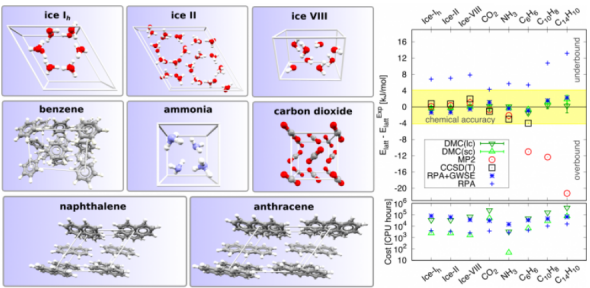
Computer simulation plays a central role in modern-day materials science. The utility of a given computational approach depends largely on the balance it provides between accuracy and computational cost. Molecular crystals are a class of materials of great technological importance which are challenging for even the most sophisticated ab initio electronic structure theories to accurately describe. This is partly because they are held together by a balance of weak intermolecular forces but also because the primitive cells of molecular crystals are often substantially larger than those of atomic solids. Here, we demonstrate that diffusion quantum Monte Carlo (DMC) delivers subchemical accuracy for a diverse set of molecular crystals at a surprisingly moderate computational cost. As such, we anticipate that DMC can play an important role in understanding and predicting the properties of a large number of molecular crystals, including those built from relatively large molecules which are far beyond reach of other high-accuracy methods. While we focus on molecular crystals, the significance of our findings extends to all classes of materials.