Atomistic details of copper oxide surfaces and surface oxidation
20th December 2015

Atomistic details of copper oxide surfaces and surface oxidation
The oxidation and corrosion of metals are issues which affect many of our everyday objects, from water pipes to electronic devices. They eventually lead to material failure and are extremely costly to businesses and individuals. These important problems have been studied for a long time. Copper is one of the most studied metals and it has become a model to understand oxidation and corrosion. In this review we show that now now have a good atomistic understanding of the physical characteristics of copper oxides and of some key processes of their formation . However a number of challenges remain. For example, the atomistic details of the nucleation of the oxide is still unknown. However we believe that recent advances in experimental techniques, bringing greater temporal and spatial resolution, along with improvements in the accuracy, realism and timescales achievable with computational approaches make it possible for these questions to be answered in the near future.
This study has been published in Surface Science Reports
Journal link:Surface Science Reports 70, 424 (2015)
Enhanced low-energy electron emission radioactive films
25th July 2015
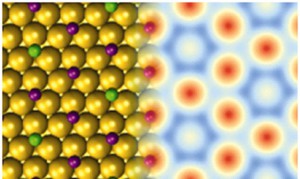
DFT structure of the radioactive film (left; I in purple and Te in green) and DFT-based simulated STM image (right). Te atoms bind stronger to the substrate than their I neighbours.
Nuclear decay is one of the most extreme processes and is central to a range of fields including energy, medicine, imaging, labelling, archaeology and sensing. Low-energy electrons from radioactivity decay are biologically active. However, it is challenging to generate them locally. In this work, a new radioactive film that enables enhanced low-energy electron emission has been synthesized. Scanning tunnelling microscopy, supported by electronic structure simulations explained the strong stabilty of the film as it underwent radioactive decay. This new radioactive film may have future applications in cancer therapies using nanoparticles.
This study has been published in Nature Materials
Journal link:Nature Materials 14, 904 (2015)
Simulating water slippage
25th January 2015
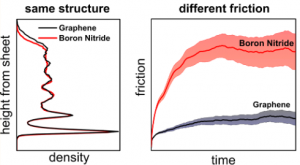
Very similar water structure VS. Very different friction
Friction is one of the main sources of dissipation. For instance, about one third of the world mechanical energy is dissipated into friction [1]. Understanding nanoscale friction at the interface between a liquid and a solid is also crucial for the development of efficient membranes for water desalination and power harvesting. Researchers at UCL in a collaboration study with Laurent Joly, from the University of Lyon, have investigated the friction properties of liquid water at the interface with graphene and with an hexagonal boron nitride sheet, using ab initio molecular dynamics for the first time. They found the striking result that the friction coefficient on boron nitride is ≈3 times larger than that on graphene although the structure of the water layers on the two sheets is almost identical.
This study (recently published in Nano Letters) has recently appeared on the Research Highlights of Nature Nanotechnology.
Journal link:Nano Lett. 14, 6872–6877 (2014)
Research Highlights in Nature Nanotechnology: http://www.nature.com/nnano/reshigh/2015/0115/full/nnano.2014.330.html
[1] Szeri, A. Z. Tribology: Friction, Lubrication, and Wear (Hemisphere, 1980).
Quantum simulation of low temperature metallic hydrogen
25th June 2013

A snapshot from the ab-initio path-integral molecular dynamics simulation where a metallic liquid phase is found at 900 GPa and 50 K. Accounting for the quantum nature of the nuclei is essential for the appearance of this low-temperature metallic phase.
The nature of dense hydrogen is a central problem in physics and its abundance, for example, in gas giants such as Jupiter and Saturn means that it is critical to our understanding of the universe. In spite of the tremendous progress made over the last 80 years, important gaps in our understanding of the hydrogen phase diagram remain, with arguably the most challenging issue being the solid to liquid melting transition at ultra-high pressures.
This study, involving a group of researchers from the Thomas Young Centre at UCL, as well as from Peking University, Cambridge and York, presents a fundamental advance in the understanding of dense hydrogen which has far reaching implications for a wide range of scientific fields.
The scientists from the four universities looked, in an international effort, at the melting of hydrogen by computer simulation of the coexistence of the solid and liquid phases, for the first time taking the quantum motion of the protons into account explicitly. The findings show a low-temperature metallic atomic liquid phase of hydrogen at pressures 900 GPa and above, down to 50K, the lowest temperature that can be reliably simulated. The existence of this low temperature liquid is associated with a negative slope of the melting line between atomic liquid and solid phases at pressures between 500 and 800 GPa. These results are highly quantum in nature, with classical simulations demonstrating completely different behaviour, with the simulations showing considerably higher melting points. This study confirms the existence of this phase in simulations and shows how the quantum motion of the protons plays a critical role in its stabilisation.
This work has been published in Nature Communications
Journal link:Nature Communications
Large variation in ice surface vacancies
25th March 2012
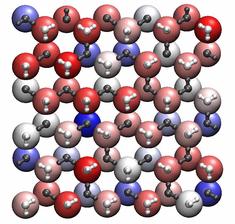
A view of the ice surface illustrating weakly, (red), intermediate (white) and strongly bound water molecules (blue). White molecules are at the external surface, grey lie sub-surface.
Ice exhibits a phenomenon known as pre-melting which was first alluded to by Michael Faraday in his ‘regelation’ experiments at the Royal Institution in the 1850’s. A liquid like layer forms at the surface of ice, but there is dispute about the temperature at which this layer first occurs.
Understanding the structure of the layer and its temperature dependence is important in the context of atmospheric heterogeneous catalysis, because the surface of ice particulates facilitate reactions of radicals and trace gases in the atmosphere.
In a recent paper in Nature Materials, TYC researchers at UCL (Matt Watkins, Angelos Michaelides and Ben Slater) in collaboration with researchers from University of Zurich, Peking University and the Chinese Academy of Sciences have discovered unexpected properties of ice at the nanoscale that relate to Faraday’s experiments. Using density functional theory calculations, they discovered each molecule is bound to the surface by a difference force, unlike most crystalline materials where each surface molecule has an identical binding energy. A fraction of the surface molecules are so weakly bound that they are easily displaced to form an overlayer, leading to less crystalline surface layers. The figure illustrates the variation in binding energy, which arises from the interaction of the water’s dipole within a geometrically frustrated array of neighbouring dipole moments.
This work has been published in Nature Materials
Journal link:Nature Materials.
Nano ice melts at -100 degrees!
25th March 2011

A partially melted ice nanoparticle at about -100 degrees Celsius
Computer simulations provide a molecule’s eye view of the melting of ice nanoparticles, predicting melting at very low temperatures.
The melting of ice is a very familiar process but its ubiquity belies its importance. It plays a central role in a wide variety of chemical processes, and is particularly relevant to environmental and atmospheric chemistry. However, despite being an everyday process, melting is not as well understood as one might have thought, particularly on the nanoscale.
Ice cubes, like those you might put in a gin and tonic melt at zero degrees Celsius, but at what temperature do ice nanoparticles (that is particles about 0.000000001 metre large) melt? An international team of researchers from the Thomas Young Centre, the London Centre for Nanotechnology, UCL Chemistry Department and Peking University set about to answer this question with computer simulation techniques and some of the most powerful computers in the UK (the HECToR and Legion Supercomputers). The answer they got is around a cool minus 100 degrees Celsius. That is, on the nanoscale the melting point of ice particles is about 100 degrees less than it is for macroscopic (everyday) ice cubes.
The strong dependence of melting temperature on particle size sounds remarkable but is not that surprising: it is due to the large surface to volume ratio of the nanoparticles and can be explained pretty well with textbook thermodynamic theories that relate melting temperatures with particle size.
For more information see the article by Pan et al. in ACS Nano.
Computer simulations revealing how salt crystals dissolve in water featured on the cover of PCCP
Computer simulations reveal in the most exquisite detail how salt crystals dissolve in water.

Figure: Cover of PCCP 7th August, 2011. Close up on salt dissolving: A snapshot along the reaction pathway as a chlorine ion leaves the corner of a salt crystal.
Salt dissolution has a resonance with scientists and non-scientists alike being a piece of “chemistry” exploited daily to inhibit the freezing or accelerate the boiling of water. Despite this key role and increased contemporary drivers from e.g. nanotechnology and the desalination industry, the mechanism of salt dissolution has however remained elusive.
To address this longstanding problem a team of researchers from the Thomas Young Centre, the London Centre for Nanotechnology, UCL Chemistry Department and SISSA, Trieste used a combination of highly accurate quantum mechanics based computer simulation approaches, sophisticated free energy sampling techniques, and some of the most powerful computers in the UK (the HECToR and Legion Supercomputers).
In stark contrast to the qualitative textbook model of salt dissolution, the mechanism arrived at from these studies is surprisingly complex, involving multiple steps and a well-defined intermediate state in which the leaving ions are partially solvated and still retain contact with the crystal. The mechanism predicted by the computer simulations can be seen at:YouTube.com !
The results of this study may prove crucial to diverse fields such as the atmospheric sciences in better understanding the desalting of aerosol solutions during freezing.
For more information see the article by Liu et al. in PCCP or www.ucl.ac.uk/ice.
A quantum theory of hydrogen bonds
Posted Jul-2011
Xinzheng Li, Brent Walker, and Angelos Michaelides
(London Centre for Nanotechnology and Department of Chemistry, University College London)
In a paper published in Proceedings of the National Academy of Sciences Xinzheng Li, Brent Walker, and Angelos Michaelides solve a 50 year puzzle about the quantum nature of hydrogen bonds.
Hydrogen (H) bonds are essential to life on earth. They are, for example, the main intermolecular interactions responsible for binding the two strands of DNA and holding together the condensed phases of water.

Figure: Squaric acid, a strong hydrogen bonded crystal, which possesses delocalised hydrogens (small grey spheres) in a strong covalently bonded framework of carbon (turquoise spheres) and oxygen atoms (red spheres). Quantum effects cause the hydrogen bonds holding this material together to strengthen.
Because H is the lightest element, it exhibits unusual quantum properties such as the ability to “tunnel” through energy barriers. These quantum properties make H-bonds highly sensitive to isotope changes. Isotopes are atoms that contain the same number of protons but a different number of neutrons. Deuterium is the most common isotope of H and when H-bonds involve deuterium rather than H something strange happens: some get longer, some get shorter and some show no change at all. This behaviour has remained an enigma since its first detection in the 1950s.
In their paper Li et al. solve this puzzle by showing that the influence isotope effects have — and in essence quantum effects — on H-bonds depends on their strength: strong H-bonds get stronger and weak H-bonds get weaker. This surprisingly simple finding is arrived at on the basis of state-of-the-art computer simulations on the UK’s largest supercomputer (HECToR) in which both the electrons and the nuclei are treated as quantum mechanical objects. It rationalises in a clear manner the seemingly conflicting results previously reported on different classes of H-bonded systems (gas phase clusters, solids including ferroelectrics, and liquids).
Detailed analysis of the underlying physics shows that the correlation arises from competition between covalent (intramolecular) bond stretching and intermolecular bond bending — different modes dominate in strongly and weakly bonded systems.
The simple rule of thumb identified in Li et al.’s work could in the future be used by experimentalists to interpret measurements on isotope effects in H-bonded liquids, ferroelectric phase transition temperatures in H-bonded crystals, high pressure phases of ice, and proton transfer probabilities in H-bonded biological materials (e.g. beta sheets and alpha-helixes).
For more information see Li et al. Proc. Nat. Acad. Sci.
Water breaks the rules: An ice-like overlayer stabilised by Bjerrum defects
Posted Feb-2011
Matthew Forster, Rasmita Raval, Andrew Hodgson
(Surface Science Research Centre and Department of Chemistry, University of Liverpool)
Javier Carrasco
(London Centre for Nanotechnology and Department of Chemistry, University College London, and Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin)
Angelos Michaelides
(London Centre for Nanotechnology and Department of Chemistry, University College London)
On many wet surfaces the first contact layer of water is not comprised of pure water but is instead a mixture of water and hydroxyl molecules, often caused by the spontaneous dissociation of water at the interface. Understanding the composition and stability of these layers is a key step in describing wetting and how surfaces respond to redox processes of importance in a variety of fields such as electrochemistry, geology, and biology. In collaboration with Matthew Forster, Rasmita Raval and Andrew Hodgson (University of Liverpool), Javier Carrasco and Angelos Michaelides (LCN and TYC) have shown that structures containing an excess of water over hydroxyl are stabilized on a copper surface by forming a distorted hexagonal network containing Bjerrum defects (in which two H atoms sit between two adjacent O atoms). This novel defect rich structure, which is contrary to what the so-called “ice rules” would predict, was identified through a combination of scanning tunneling microscopy (STM) experiments in Liverpool and density functional theory (DFT) calculations in London. Careful analysis reveals that the overlayer forms because it maximizes the number of strong bonds formed by water donation to OH. As a result this work also emphasizes that maximizing the number of hydrogen bonds per molecule is not necessarily a useful way to identify stable structures as previously assumed. It is also shown in the paper that the Bjerrum defects provide uncoordinated OH groups able to hydrogen bond multilayer water and nucleate growth. This suggests that defects will facilitate ice nucleation, one of the most important everyday processes of particular relevance to atmospheric chemistry and cloud formation.
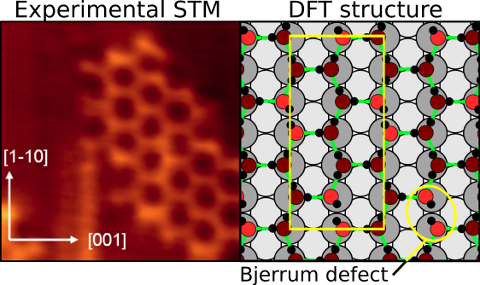
On the left is an STM image (Hodgson and co-workers, University of Liverpool) showing a distorted hexagonal arrangement of overlayer molecules. The theoretical (DFT) structure (right) shows the precise geometric arrangement of this overlayer with the Bjerrum defects highlighted.
Read more in Forster et al., Phys. Rev. Lett. 106, 046103 (2011)
To wet or not to wet? Dispersion forces tip the balance for water-ice on metals
Posted Jan-2011
Javier Carrasco, Biswajit Santra, Jiri Klimes, and Angelos Michaelides
Ice formation on metal surfaces plays a fundamental role in fields as diverse as the atmospheric sciences, geology, and biology. A prerequisite to understanding these wide and varied phenomena is establishing how the water molecules are arranged at the water-metal interface. Density functional theory represents the state-of-the-art theoretical methodology to tackle efficiently this problem. However, if adsorption energies with the most commonly used exchange-correlation functionals are to be believed, none of the low temperature experimentally characterized ice-like wetting layers are thermodynamically stable. Since the typical density functionals used to date do not account properly for van der Waals (vdW) dispersion forces, it would of course be timely and important to know what role vdW forces play in water adsorption on metals. In a recent paper published in Physical Review Letters, Carrasco et al (Phys. Rev. Lett. 106, 026101 (2011)) show that when vdW interactions are accounted for, this discrepancy between experiment and theory can be reconciled. The resolution of this long-standing anomaly is demonstrated on one of the most well-characterized wetting layer structures (water on Cu(110)) and on the most widely investigated (water on Ru(0001)).

Standard density functionals predict that water-ice should not wet metals, but rather form three dimensional non-wetting ice crystals. Accounting for dispersion forces rectifies this problem and produces a result in agreement with experiment.
Read more in Carrasco et al., PRL 106, 026101 (2011)
Quantum nature of the proton in water-hydroxyl overlayers on metal surfaces
Posted mid-2010
Xin-Zheng Li, Matthew I. J. Probert, Ali Alavi, and Angelos Michaelides
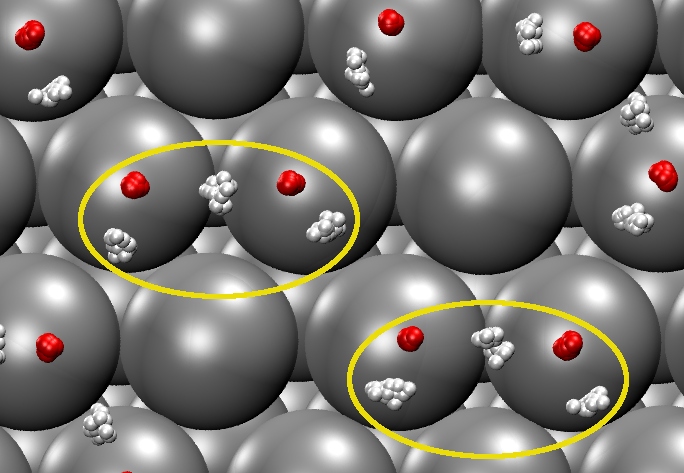
On many surfaces the first contact layer of water is not comprised of pure water but instead of a mixture and water and hydroxyl molecules. Using ab initio path integral molecular dynamics (PIMD) we show that these overlayers on transition metal surfaces exhibit surprisingly pronounced quantum nuclear effects. The surfaces serve to reduce the classical proton transfer barriers within the overlayers and, in analogy to ice under high pressure, to shorten the corresponding intermolecular hydrogen bonds. Depending on the substrate and the intermolecular separations it imposes, the traditional distinction between covalent and hydrogen bonds lost partially (e.g. on Pt(111) and Ru(0001)) or almost entirely (e.g. on Ni(111)). The above figure shows an example from the PIMD simulation on Pt(111) at 160 K where due to the quantum fluctuations of nuclei the distinction between water and hydroxyl lessens considerably and stable H3O2 groups (indicated by the yellow circles) forms in which the proton is equally shared by the oxygen atoms. Reducing the substrate lattice constant increases the magnitude of these quantum effects. These results suggest that such systems provide an excellent platform to systematically explore the magnitude of quantum nuclear effects in hydrogen bonds.
Read more in Li et al., PRL 104, 066102 (2010)
A novel one dimensional ice structure built from pentagons has been discovered…
Posted mid-2009
Javier Carrasco, Angelos Michaelides, Matthew Forster, Sam Haq, Rasmita Raval, and Andrew Hodgson

Heterogeneous ice nucleation plays a key role in fields as diverse as atmospheric chemistry and biology. Ice nucleation on metal surfaces affords an opportunity to watch this process unfold at the molecular-scale on a well-defined, planar interface. A common feature of structural models for such films is that they are built from hexagonal arrangements of molecules. Here we show, through a combination of scanning tunneling microscopy, infra-red spectroscopy, and density-functional theory, that ca. one nanometer wide ice chains that nucleate on Cu(110) are not built from hexagons, but instead are built from a face sharing arrangement of water pentagons. The pentagon structure is favored over others because it maximizes the water-metal bonding whilst at the same time maintaining a strong hydrogen bonding network. It reveals an unanticipated structural adaptability of water-ice films, demonstrating that the presence of the substrate can be sufficient to favor non-hexagonal structural units.
Read more in Carrasco et al., Nature Mater. 8, 427 (2009)
Related press stories: New Scientist ; Chemistry World ; Fox News
The Ice Surface is Superchilled: First principles simulations predict that the ice surface is proton ordered before the onset of pre-melting
Posted January 2009
Ding Pan, Li-Min Liu, Gareth A. Tribello, Ben Slater, Angelos Michaelides and Enge Wang
We all know that ice becomes slippery some 20-40 K below its bulk melting point. Less is known, however, about the surface of ice at lower temperatures such as those experienced by ice crystals in the upper atmosphere. Here, Pan et al. show through first principles electronic structure simulations that although bulk ice is a proton disordered solid, at the surface, protons order. Electrostatic repulsion between protons at the surface cause them to line up, effectively making the surface superchilled. This insight into the ice surface is likely to have implications for the equilibrium crystal shape of ice crystals or catalytic reactions which take place on their surfaces.
Read more in Pan et al. Phys. Rev. Lett. 101, 155703 (2008)
Density Oscillations in a Nanoscale Water Film on Salt: Insight from Ab Initio Molecular Dynamics
Posted 18 December 2008
Limin Liu, Matthias Krack, and Angelos Michaelides
The salt-water interface is one of the most important and common on earth, playing a prominent role in disciplines such as atmospheric science and biology. Despite the apparent simplicity of such interfaces, arguably the most fundamental question of what the nature and structure of the liquid water/salt interface is under ambient conditions remains unclear. Here we address this issue with an ab initio molecular dynamics simulation of a nanoscale liquid water film on NaCl. A pronounced layering is observed in the film, with the density exhibiting a damped oscillatory behavior in the direction of the surface normal. In addition, water molecules in the contact layer are preferentially adsorbed at specific adsorption sites, involved in about 20% fewer hydrogen bonds with each other, and carry considerably reduced dipole moments compared to bulk liquid water.
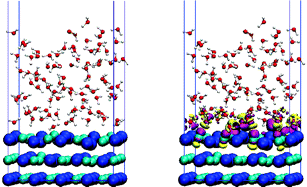
For more information see: J. Am. Chem. Soc., 130 (27), 8572–8573, 2008. 10.1021/ja8014296