Welcome to the Tips, Videos and Knowledge Base page, where all our tutorial videos can be found. Beneath the video list are some tips and tricks to help you with your NMR analysis, followed by our new knowledge base where some of the more common questions we receive are answered.
NMR Facility Tutorial Videos
These short training videos will cover most of the common problems and questions we encounter on a daily basis. We recommend downloading .avi videos to your computer or device and playing it from there for best performance. Later videos are available on the University Streaming Service NMR Facility collection at https://www.sms.cam.ac.uk/collection/3308262 . Please send any questions or queries about these videos to the NMR Facility team at nmr@ch.cam.ac.uk.
NMR Facility Video 001 - Introductory Video - Introduction to the NMR Facility
Video Tutorial 002 - Filling in a Sample Submission Form - Filling in a Submission Form.avi Slightly out of date as we have changed to online submission only due to the COVID-19 pandemic, and redesigned the form slightly, but the essence of the information given is the same.
Video Tutorial 003 - Preparing your NMR Service sample in a 5mm NMR tube - Preparing your sample in a 5mm tube.avi
Video Tutorial 004 - Open-access NMR training 1 - General Principles and Safety when using open-access NMR
Video Tutorial 005 - Open-access NMR training 2 - Submitting a sample to NMRKiosk
Video Tutorial 006 - Pronunciation of our NMR instrument names
NMR Service Tips and Tricks
Here are some tips and tricks to make your life easier and solve some of the common problems encountered when submitting samples to the NMR Service or using one of the spectrometers.
Common impurities charts
Don't get mistified by signals of unknown origin in your NMR spectra. View the very helpful Sigma-Aldrich charts of common impurities in your 1H and 13C spectra in various solvents by clicking on the link below:
Preparing your sample - Now available as a video tutorial (see above) When making up your sample try to avoid floating impurities or other solid material in your NMR tube. These will affect the quality of your spectrum. If necessary, filter your solution before putting it in the NMR tube. Aim to get about 0.7ml of solution in your NMR tube. The individual vials of solvents that the NMR Service sell are 0.75ml, sufficient for one sample at the correct depth in the tube. Too little solvent means that the air-liquid boundary may fall in the area of the probe coils leading to field inhomogeneity. Too much solvent means your sample concentration is lowered leading to a requirement for a longer acquisition time.

NMR tubes - Your NMR tubes should be straight, free from scratches, and a minimum of 17.5cm in length. This will mean that you can use your tube in any of the systems with a sample changer. Short tubes may fail to be picked up by the sample changer, which could stop the automatic run. You should always use Wilmad 528PP tubes in the departmental spectrometers - other makes of tube may slide too easily in the spinners, break more readily and not be manufactured to such fine tolerances leading to inhomogeneity and poor spectra. The NMR Service reserve the right to remove any non-standard tubes from the open-access spectrometers. You should take care when placing a cap on your tube, especially if the top of the tube is chipped. We sometimes have second-hand Wilmad 528PP tubes for sale at half the normal price - please ask in B28. Tubes that have been gathered from the open-access spectrometer rooms and have gone uncollected for two weeks are available on a first-come first-served basis, usually from 8am on Tuesday mornings.
Inserting your tube into a spinner - You should take care when you insert a tube into a spinner (turbine). Grip the tube towards the bottom and gently insert into the spinner. Do not exert too much force - you may find that rotating the tube as you push it helps. Keep your hand near to the spinner as you push the tube further into it. Gripping the tube at the top and attempting to insert it into a spinner can create unnecessary forces which may cause the tube to snap with the potential for injury. When using a ceramic spinner for variable temperature work you should take extra care as they tend to be tighter than the normal spinners.

Breakages - We are all human, and sometimes things go wrong. If you break a tube in an NMR room, please try and clean up as much as possible, collecting the broken glass and disposing of it in a safe manner. If you break a tube when it is in a spinner, please bring the spinner to the NMR Service in B28. We have a tool for removing the glass from the spinner, and can clean it and return it to service. Please do not throw the spinners away - they are very expensive to replace. If a tube breaks inside the magnet, you should stop work immediately and inform the NMR Service. If this occurs outside normal working hours, then you should leave a large note on the instrument to warn others not to use it. Please email or leave a note for us - we will endeavour to clean inside the magnet and probe and restore the instrument to service as quickly as possible.
At the spectrometer - When you have inserted your sample into the magnet it is good practice to load the default shimset. The previous user of the instrument may have been working with an unusual sample and the shims may be a long way from their default position. This could make your sample difficult to lock and shim. Type rsh and select the shimset named current or similar. This will provide a good starting point for shimming your sample with Topshim. You should not assume that the probe is tuned correctly either. Check the tuning using atma (or by typing wobb and manually adjusting the knobs on the underside of the probe on non ATM equipped probes). If you have any problems with locking and shimming your sample, or tuning the probe then please see us in B28 as we will be able to help you get the best possible result.
Variable temperature NMR - When running experiments at very high or low temperatures you should use a ceramic spinner. You should also use the bore gas system which helps maintain the bore and o-rings in the magnet nearer to room temperature. This is operated from the top of the magnet. Please see the NMR staff in B28 if you do not know how this works. You should use the default configuration files when changing between temperatures, as these have heater power and gas flow calibrated. If you need a different temperature, select the nearest temperature file, and just change to the exact temperature required manually. The BBO Smart Probe 500MHz spectrometer (Glengrant) has a BCU chiller unit that can lower the temperature to -80C - controlled as normal using the edte window. The old style chiller units will lower the temperature to -5C, below this you will need to use the liquid Nitrogen tank and apparatus (with compatible probes). See the NMR Staff if you have not used this before. When using the TCI 500 spectrometer (Glenlivet), please be aware of the upper temperature limit of the probe which is 45C. Increasing the temperature beyond this limit will trigger the Cryoprobe to warm itself up, thus ruining your experiment. Please allow extra time to return the probe to the default temperature when booking slots on the instruments. For sessions of a number of hours at elevated or lowered temperature, it could take up to 30 minutes to re-equilibrate at the default temperature.
Filling in a submission form - Now available as a video tutorial (see above) - The sample submission form (available on the NMR Home page) helps us decide on the length of experiments we will run. Please take a little time to fill it in accurately and correctly. The sample weight and molecular weight of your sample are the most important pieces of information. The difference between 2mg and 0.5mg could be the difference between hundreds and many thousands of scans for a carbon experiment. If you don't know the exact molecular weight then make a very rough calculation to the nearest hundred. Don't forget to fill in your solvent details - we don't like having to sniff your tube to find out what it is! Try to write clearly, especially your name, telephone number and email address - we sometimes need to contact you before running your sample. Your personal code should relate to an entry in your lab book, and this is what we will name the experimental files prefixed by your group initials. For example jrn-abc1-25-prep. Please include the structure if you know it, it does help us to decide if the NMR experiment is successful or otherwise.
Salty (Ionic) samples - These types of samples often cause problems when they are very ionic. In cryoprobes especially they can be difficult to tune and result in broader peaks. To counter this you can move the sample further away from the NMR coils. This is achieved by using a 3 or 4mm tube and the special spinners available from B28. By using the same amount of sample with the reduced volume of solvent you can increase the number of nuclear spins in the active volume thus helping to claw back some of the sensitivity lost by having the sample further away from the coils.

Topspin installation and use - TopSpin 4.x is now free for academic use. For information and assistance with this software please see our home page. We have an installation guide for TopSpin 4.x and Duncan has also written a guide to processing using TopSpin which is very informative.
NMR Facility Knowledge Base
In this new section of our website we aim to answer some of the common questions that we receive in person and by email on a regular basis as each new intake of researchers get to grips with NMR.
1. How the heck do these Accuratus keyboards work??
2. How do you use this booking system? How can I set one up?
3. Quantitative NMR - a quick overview
4. TopSpin - Changing the default plot settings
5. TopSpin - How to plot multiple spectra
6. Don't be DOSY - check out our quick guide to setting one up here!
7. TopSpin data won't open on my new or upgraded Mac
8. Can you help me set up a selective NOESY or ROESY?
9. I need to use a solvent that isn't on the open-access instruments!!
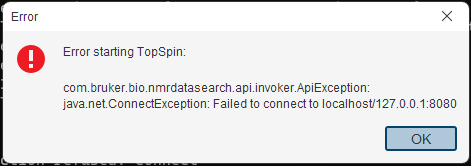
10. Help, TopSpin won't start anymore, it just shows this error message!
11. At what time did my data actually acquire?
12. Getting to grips with accurate water suppression.
13. How to shift spectra independently when using multiple display (.md) in TopSpin
14. Fluorine and the curiosity of its long range couplings to 1H
Our new Accuratus glass keyboards are fantastic for sanitising in the current COVID-19 climate, but they can be a little tricky to get used to. Firstly there is a quick instruction guide put together by Duncan available here. Secondly there are a few tricks to getting it to work when it appears to have given up the ghost, usually as a consequence of someone poking the wrong key or button!!
Symptom: Nothing happens.......
Are the lights on? If not, try each of these in turn until something does!!
- Push 'Wake'
- Check the keyboard isn't locked. If it is, hold the 'Lock' button for 3 seconds until it unlocks
- Turn the keyboard power on with the power switch at the back
- Plug the white USB-C cable into the back of the keyboard
The lights are on, but nothing happens
- Toggle the connection to '2.4'
- Plug the USB cable in and toggle to USB
Symptom: The mouse doesn't work
- Toggle the trackpad to on
All the controls can be found in the PDF guide link above.
Clustermarket Manuals and Guides are all available at their website and the link is here!
It is quite intuitive to use as a basic booking system but if you are setting it up for a large facility with different groups of users with different training records and different requirements it does take a little learning. Don't learn on the job like Andrew, just download the manual.....![]()
Questions about using NMR quantitatively crop up all the time! Duncan has a pretty stock standard answer which serves as a quick overview of the perils and pitfalls of relying on your NMR spectrum to give a quantitative answer!
TopSpin - plot settings and how to change them! Have you cursed and sworn at the dodgy plot editor TopSpin has saddled you with? You're not the only ones! Zhang-He Goh (Matt Gaunt's PhD candidate 2020) has spent many hours getting TopSpin to produce beautiful output the way he wants with some help from Duncan. Here we summarise how to change the plot setttings to output the things you want and not the things you don't!!
(Note: TopSpin does have all its manuals ‘built in’. Just click the question mark in the top right of the main TopSpin window. The one you want here is the ‘data publishing’ one.)
So clicking the spectrum will select it, you can then manipulate it. Then there’s a nearly invisible arrow you click on to go back to the page where you can add new elements and save the layout. The UI design for this is terrible…![]()

Create the layout you want, then save it to a new file. Eg - 1h-red-pp.xwp

For bonus points you can make a macro to load this layout into your dataset, so when you click plot, it appears just how you want!
-At the command line type :-
edmac 1h-red (where the red type signifies that 1h-red is the name of the macro)
Paste the following lines into it :-
Save it. Now you can type 1h-red to load a defined plot layout in, and set a defined plot region. So finer points on usage….
Run the macro 1h-red
Right click on the spectrum in the main topspin windows and chose ‘restore display region from params’
Type plot
The correct region should appear in the plot layout. If it doesn’t, it's due to the underlying layout ‘automation actions’ being incorrectly defined. I do not find the automation actions work in a rigorously consistent fashion!
Then Zhang-He turned his attention to the annoying file path that appeared right across his stacked plot output. Very irritating as you can see:
But with a little searching the button to remove the file path can be found in the toolbar and in the image below Zhang-He has highlighted it in blue.

Plotting multiple spectra on TopSpin can be a right pain in the arse - let's not beat about the bush here!!! Duncan has spent many hours trying to get to grips with it and he shares his knowledge in the document you can download right here! This is currently a beta version and is still being worked on from time to time but it has been used successfully by some researchers who want to master the vagaries of TopSpin before it is consigned to history!!
Setting up your DOSY experiments should follow a set protocol every time to make sure you get the reproducibility that means the result can be trusted! Don't let variations in experimental parameters including not knowing the T1 value, and unwanted convection leave your results all over the place. This is a quick step by step list of what you need to do:
-
Do an rpar proton.std all
-
Log outSet the correct pulse program with pulprog zg
-
Set dummy scans to zero with ds=0
-
Set to scan only once by typing ns=1
-
Optimise the 90 degree pulse - find the 360 degree pulse and divide by 4 is the best way as the 360 is a null
-
Change the pulse program: type pulprog t1ir
-
Create a list of delays by typing edlist. Call it something easy to remember. Define maybe 8 entries in steps from 0.01s up to close to the expected T1
-
Set VDLIST in the acquisition parameters to the name you used in step 7
-
Change the experiment to a 2-Dimensional one
-
Change dummy scans to 4 (ds=4) and the number of scans to 8 (or a multiple of 8) using ns=8
-
Set F1 TD to the number of entries in your list from step 7
-
Type d1=xx where xx is 5x the longest delay in your list
-
In the processing parameters set F1 SI to the number of entries (or the next power of 2 above) in your list
-
Let's go - do rga then zg
-
Type xf2
-
Phase correct the results
-
Finally fire up the Dynamics Centre and process the results there
TopSpin won't work on my new or upgraded Mac - is an email we receive quite frequently and the answer is usually that the Mac OS X has decided to take exception to the non-native program and where you put the data that you have collected on our instruments. As a Mac user (for his sins), Duncan has encountered more of these errors than he cares to remember and always recommends the following:
The newer versions of OS X are ‘touchy’ about where data goes. I create a directory called nmr-in in my home directory, so /Users/Duncan/nmr-in and put data in there.
The two versions of OS X have entries in the TopSpin release notes that you can read by clicking the ? at the top left corner and choose manuals > release letter.
Here are the entries for Catalina and BigSur (why they have silly names I will never understand - Andrew :-) ):
The macOS version 10.15 “Catalina” introduced several security restrictions. Apps must now request permission to access specific parts of your file system. Unfortunately, the Apple implementation is not well documented and does not take into account all possible software architectures. This can affect the function of TopSpin. Viewing and processing data stored on a local disc works as expected. However, viewing and processing data from mounted external drives may fail. In most cases, a simple workaround helps:
Open the Terminal application (found in the directory /Application/Utilities/) and start TopSpin from command line (e.g., /opt/topspin4.0.8/ topspin).
This is valid for all TopSpin versions, not just 4.1.3.
The latest macOS release 11.0.1 "Big Sur" introduced several low-level changes, which affect the compilation of AU programs in TopSpin. In order to be able to compile and link AU programs, an installation of Apple’s Xcode or Xcode command line tools is now required. When installing TopSpin on macOS, the installer will offer a choice to install the required tools. As an alternative solution, the Xcode command line tools may also be installed from the App store at a later time.
I just need a selective NOESY or ROESY, how do I do that? This is a question that we occasionally get when people don't really need to run a whole great 2D through space correlation experiment if they have a specific peak in mind for the irradiation. It saves a lot of spectrometer time and costs a lot less therefore, but you need to remember to set it up specifically for the peak in question each time. Here's the workflow:
- integrate the peak(s) to be irradiated – do save as reg and save as intrng
- go to the Aquire > Options > set up 1d selective experiments
- choose Noesy or Roesy
- choose an appropriate mixing time (this is molecular weight/correlation time dependent) For Noesy typically </= 1s and for ROESY <0.3s (Roesy uses a spin-lock, which has a limit before it fries things, and that should be reserved for foodstuffs only!!)
- Note: don't copy a previous experiment and just move the irradiation frequency. Each time you run the routine it calculates and creates a shaped pulse which is particular to the integral region you have saved - thus ppm and width of peak are important. To compare between similar samples you need to use the same mixing time/spin-lock time.'
I need to use a solvent that isn't in the NMRKiosk list on the open-access instruments!! This is a question that we also get occasionally for solvents such as Nitromethane or Dimethylformamide. This is awkward to set up, but not impossible (we have to modify a database using an old version of Access). However, there is a work-around that is simple!

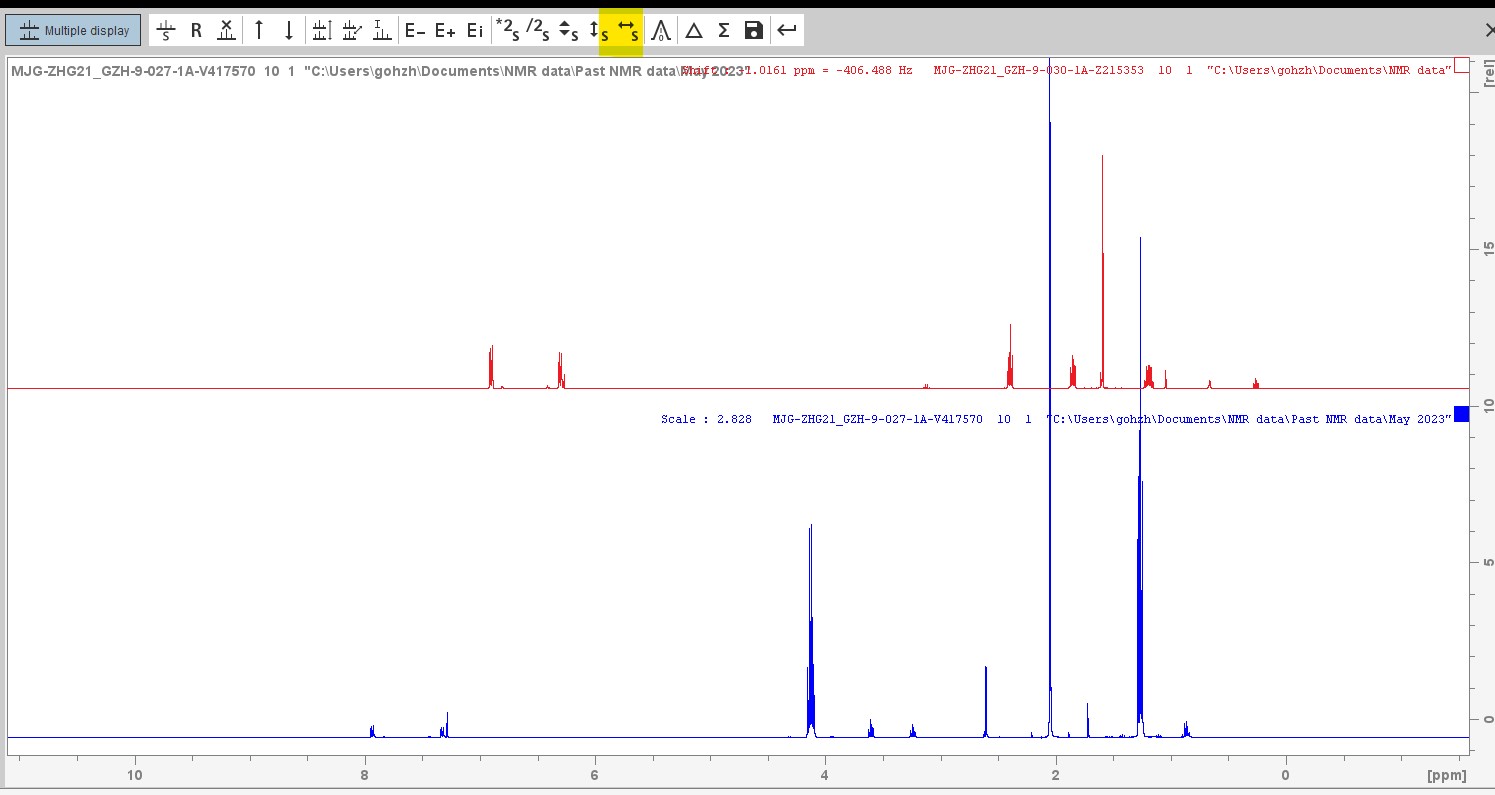
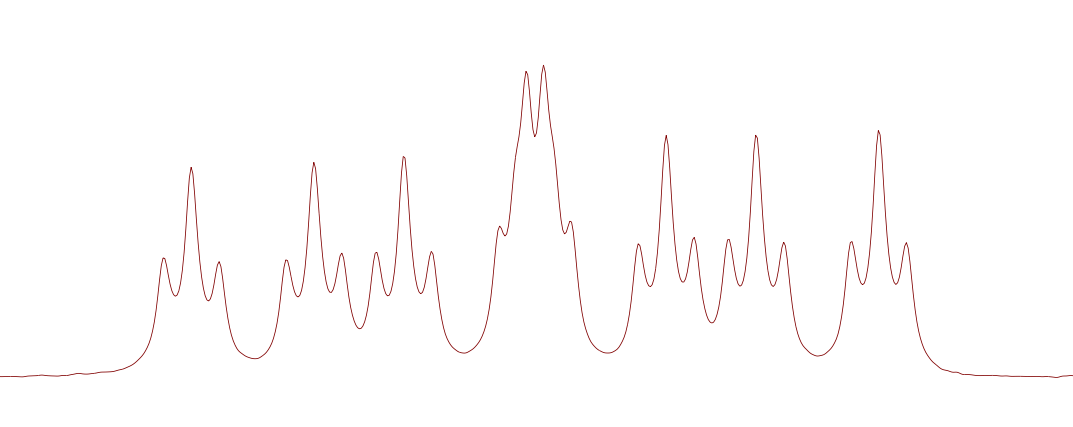
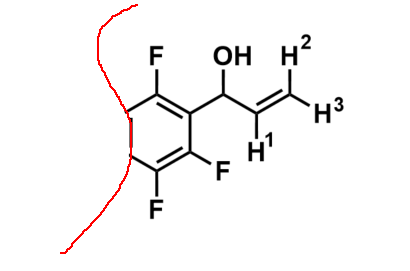 remarked that the non-fluorinated derivatives of this structure would produce the expected ddd multiplet from the coupling of H1 to the vicinal H2 and H3 and a proton on the benzene ring where one of the ortho-fluorines are situated. Surely the 5 bond coupling constant from H1 to these ortho-fluorines was far too small to observe! But what else could it be that produces this 1.4Hz 5J(H-F)?? Could we convince him of a reason why this could be possible. With that it was over to Pete to come up with something plausible. Here is his answer..........
remarked that the non-fluorinated derivatives of this structure would produce the expected ddd multiplet from the coupling of H1 to the vicinal H2 and H3 and a proton on the benzene ring where one of the ortho-fluorines are situated. Surely the 5 bond coupling constant from H1 to these ortho-fluorines was far too small to observe! But what else could it be that produces this 1.4Hz 5J(H-F)?? Could we convince him of a reason why this could be possible. With that it was over to Pete to come up with something plausible. Here is his answer.........."Couplings to fluorine can often be somewhat large (and sometimes strange - for example – 3J(F-F) in -CF2-CF2- systems is close to zero). So a five bond coupling of 1.4Hz as you see here is not very surprising. You can sometimes see really large long range couplings. Referred to as “through space”, but really due to overlap of the fluorine nonbonding electrons with a bonding orbital (eg of a CH bond), due to spatial constraints."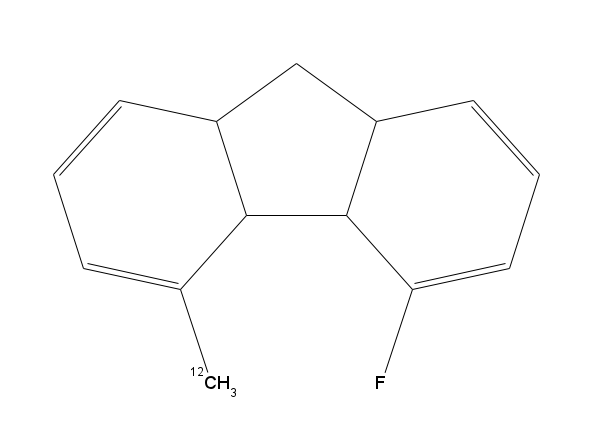
Pete goes on to offer the example of the molecule shown in image 3 on the right of the page. You really would not imagine there to be a cat in hells chance of seeing this 6 bond C-H coupling to the methyl protons. But not only is it visible, it comes in with a 6J(H-F) of 8.3Hz would you believe! To prove his point, if you reacquired the 1H NMR spectrum of the molecule in image 2, but this time with Fluorine decoupling, you remove the triplet element of the multiplet and all of a sudden it's just a boring ddd instead!! NMR is often perplexing at time but with fun examples like this at hand, it is also very interesting and rewarding!