Theory
DFT calculations for battery and fuel cell materials
We use theoretical simulations to support and guide our experimental research. An important part of the calculations done in the Grey group is related to the determination of phase energetics and NMR chemical shifts in order to interpret NMR spectra in a very detailed way. Our work includes the calculation of NMR parameters such as hyperfine shifts for paramagnetic materials as well as investigations of materials phase transitions. Current projects include investigations of LiFePO4, LiMnO2, and NaMnO2, and doped diamagnetic perovskites for application as intermediate temperature SOFC electrolytes, such as LaMgxGa1–xO3–(x/2), BaYxSn1–xO3–(x/2) and yttrium-doped BaZrO3. We have also extended calculations of hyperfine shifts to paramagnetic SOFC electrode materials such as La2NiO4+d and transition metal doped CeO2–x.
List of selected publications:
-
"Spin-transfer pathways in paramagnetic lithium transition-metal phosphates from combined broadband isotropic solid-state MAS NMR spectroscopy and DFT calculations," RJ Clément, AJ Pell, DS Middlemiss, FC Strobridge, JK Miller, MS Whittingham, L Emsley, CP Grey, G Pintacuda - J. Am. Chem. Soc. (2012) 134, 17178. (DOI: 10.1021/ja306876u)
-
"Mapping structural changes in electrode materials: Application of the hybrid eigenvector-following density functional theory (DFT) method to layered Li0.5MnO2", ID Seymour, S Chakraborty, DS Middlemiss, DJ Wales, CP Grey, Chem. Mater. (2015) 27, 5550. (DOI: 10.1021/acs.chemmater.5b01674)
-
"Defects in doped LaGaO3 anionic conductors: linking NMR spectral features, local environments, and defect thermodynamics." F Blanc, DS Middlemiss, Z Gan, CP Grey - J. Am. Chem. Soc. (2011) 133, 17662. (DOI: 10.1021/ja2053557)
DFT calculations and lattice simulations for supercapacitors
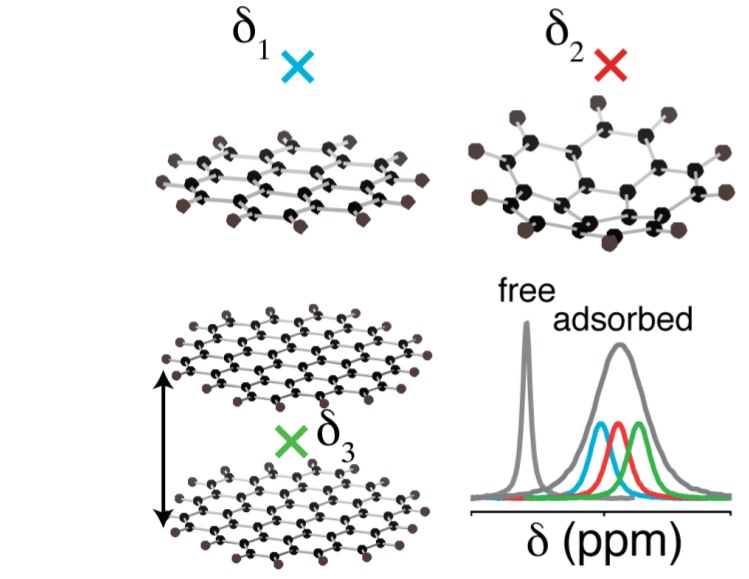 One other area of theoretical research is turned towards the understanding of ring current effects in disordered porous carbons. Indeed, NMR is increasingly being used to study the adsorption of molecules in these materials, a process which underpins applications ranging from electrochemical energy storage to water purification. We have carried out DFT calculations of the nucleus-independent chemical shift (NICS) near various sp2 hybridised carbon fragments to explore the structural factors that affect the resonance frequencies observed for adsorbed species. The carbon domain size, the pore size, as well as any curvature in the sheets all have a significant effect on the resonance frequencies calculated for adsorbed species.
One other area of theoretical research is turned towards the understanding of ring current effects in disordered porous carbons. Indeed, NMR is increasingly being used to study the adsorption of molecules in these materials, a process which underpins applications ranging from electrochemical energy storage to water purification. We have carried out DFT calculations of the nucleus-independent chemical shift (NICS) near various sp2 hybridised carbon fragments to explore the structural factors that affect the resonance frequencies observed for adsorbed species. The carbon domain size, the pore size, as well as any curvature in the sheets all have a significant effect on the resonance frequencies calculated for adsorbed species.
In ongoing work, we hope to probe the structure and dynamics of electrochemical double layers by combining these NMR calculations with lattice simulations. The idea is to use a coarse-grained model to predict NMR spectra of species diffusing in a porous network. The lattice model includes dynamical echange between adsorption sites and is parametrised using input from molecular simulations, such as NICS from DFT calculations and adsorption profiles from molecular dynamics simulations.
In our most recent work, we have used our NMR calculations and lattice simulations to probe local order in amorphous porous carbons. The simulations allow an estimation of the sizes of the hexagonally bonded carbon domains. By combining these studies with other techniques such as pair distribution function (PDF) analysis and Raman spectroscopy, it is possible to study how the carbon structure is affected by the synthesis temperature.
List of selected publications:
-
"New Insights into the Structure of Nanoporous Carbons from NMR, Raman, and Pair Distribution Function Analysis" AC Forse, C Merlet, PK Allan, EK Humphreys, JM Griffin, M Aslan, M Zeiger, V Presser, Y Gogotsi, and CP Grey Chem. Mater. (2015) 27, 6848. (DOI: 10.1021/acs.chemmater.5b03216)
-
"Lattice simulation method to model diffusion and NMR spectra in porous materials", C Merlet, AC Forse, JM Griffin, D Frenkel, CP Grey J. Chem. Phys. (2015) 142, 094701. (DOI: 10.1063/1.4913368, arXiv link).
-
"Ring current effects: factors affecting the NMR chemical shift of molecules adsorbed on porous carbons" AC Forse, JM Griffin, V Presser, Y Gogotsi, CP Grey J. Phys. Chem. C (2014) 118, 7508. (DOI: 10.1021/jp502387x)
DFT calculations for chemical looping materials
 The Materials Project is a database which contains structural, electronic and energetic data for over 50 000 inorganic compounds. The ground state energies and band structures of these compounds are calculated using density functional theory (DFT), and these calculations can be used to predict the physical properties of materials. In the group we have focussed on using the Materials Project database to predict the thermodynamics of various gas-solid reactions important in areas such as chemical looping (where oxygen is supplied in industrial processes via a solid carrier) and carbon capture. Using the in-built software packages of the Materials Project, we can simulate open phase diagrams of binary and ternary systems including a gaseous component (oxygen or carbon dioxide), and predict the partial pressure and temperature at which different phases within the system will react. This information can be gleaned for thousands of materials to screen for compounds with the desired physical properties, allowing us to rationally target certain compounds as candidates for novel industrial processes.
The Materials Project is a database which contains structural, electronic and energetic data for over 50 000 inorganic compounds. The ground state energies and band structures of these compounds are calculated using density functional theory (DFT), and these calculations can be used to predict the physical properties of materials. In the group we have focussed on using the Materials Project database to predict the thermodynamics of various gas-solid reactions important in areas such as chemical looping (where oxygen is supplied in industrial processes via a solid carrier) and carbon capture. Using the in-built software packages of the Materials Project, we can simulate open phase diagrams of binary and ternary systems including a gaseous component (oxygen or carbon dioxide), and predict the partial pressure and temperature at which different phases within the system will react. This information can be gleaned for thousands of materials to screen for compounds with the desired physical properties, allowing us to rationally target certain compounds as candidates for novel industrial processes.
High Performance Computing Resources
- Odyssey - Computing Cluster owned by Prof Stuart Althorpe and Prof Clare P. Grey;
- Darwin - University of Cambridge Supercomputing Service;
- Archer - UK National Supercomputing Service;
- Nano - Center for Functional Nanomaterials (CFN) Supercomputing Service at Brookhaven National Laboratory.