Computational Studies
We work very closely with computational chemists and physicists to benchmark simulation against experiment. Where successful, simulations can "fill in the gaps" between experimentally observable species. We have successfully used computer simulations to predict the forced unfolding pathway of a titin domain and bench-mark the simulations with experiment. We have also used experimental protein engineering data as constraints in molecular dynamics simulations to understand the structure of protein folding transition states.


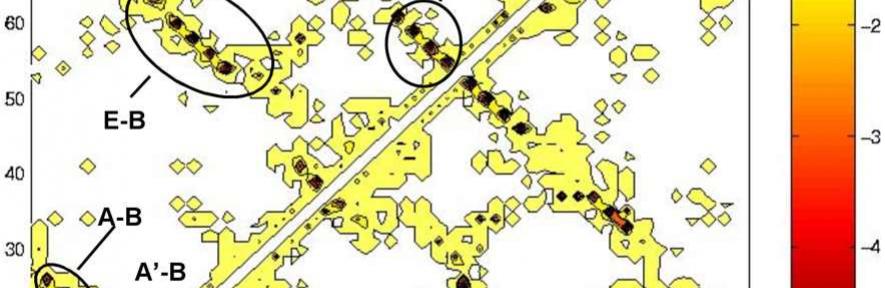
Interactions in the transition state ensembles of Ti I27. Energy maps in the equilibrated native state (upper part of the matrix) and the transition state ensemble (lower part of the matrix) for (A) TSEH and (B) TSEL. Energies are in kcal/mol.
Selected publications
- Geierhaas, C. D., Salvatella, X., Clarke, J. & Vendruscolo, M. (2008). Characterisation of transition state structures for protein folding using 'high', 'medium' and 'low' {Phi}-values. Protein Eng. Des. Sel. 21, 215-222.
- Geierhaas, C. D., Best, R. B., Paci, E., Vendruscolo, M. & Clarke, J. (2006). Structural comparison of the two alternative transition states for folding of TI I27. Biophys J. 91, 263-275.
- Ng, S. P., Rounsevell, R. W. S., Steward, A., Geierhaas, C. D., Williams, P. M., Paci, E. & Clarke, J. (2005). Mechanical unfolding of TNfn3: the unfolding pathway of a fnIII domain probed by protein engineering, AFM and MD simulation. J. Mol. Biol. 350, 776-789.