In the macroscopic world, we would never expect that if we were to shake a box containing parts of a model aeroplane, a perfectly assembled aeroplane would pop out. At the microscopic scale, however, this sort of behaviour is not quite as uncommon: for example, many viruses assemble their capsids spontaneously, without any external energy inputs. Moreover, artificially constructed materials that can spontaneously self-assemble into their target structures have also been studied extensively, and some amazing structures have been shown to form from simple building blocks: from closed clusters to complex crystals, liquid crystals and even quasicrystals.
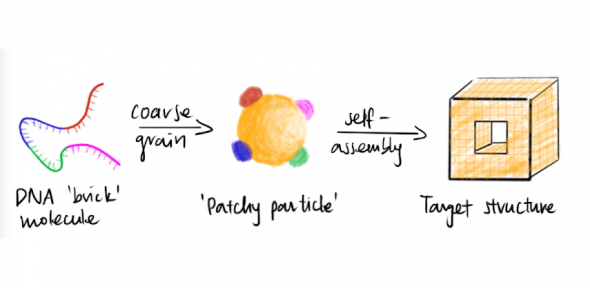
We develop simple models to try to understand complex self-assembly. In particular, DNA offers a very exciting possibility in nanotechnology: because of the specificity of its (Watson–Crick) pairings, bonding between building blocks can be carefully controlled and intricate structures that spontaneously self-assemble can be designed. However, the underlying microscopic mechanisms are not always clear, and computer simulations can help both to elucidate and to visualise them. One particular challenge in studying such self-assembly in simulation is that the structures are extremely large, containing many thousands of base pairs, making all-atom simulations intractable. Coarse-grained potentials provide a simplified picture of the system that makes the computation tractable, but should involve sufficient detail to allow the basic underlying physics of the property under investigation to be captured.
For example, we have used a variety of advanced simulation and theoretical techniques to demonstrate that DNA-brick self-assembly is rate-limited by nucleation, which has important implications for designing optimal self-assembly pathways. Given how simple the model is, it is perhaps surprising how successfully it can describe the behaviour of different aspects of the self-assembly process: in addition to rationalising the competition between nucleation and growth, which explains why DNA bricks systems can successfully self-assemble at all, the model has allowed us to design nucleation pathways and understand the fine details of the microscopic nucleation pathways, including making explicit links to experiments.
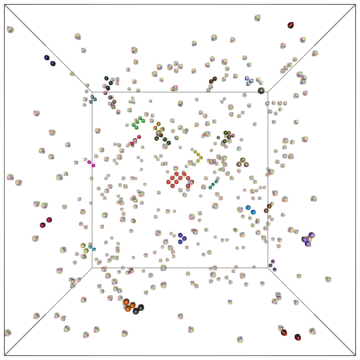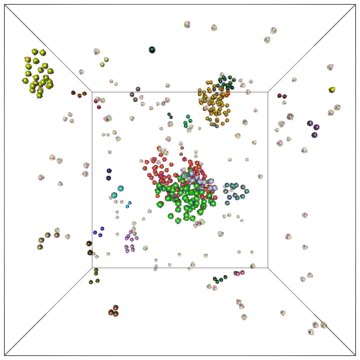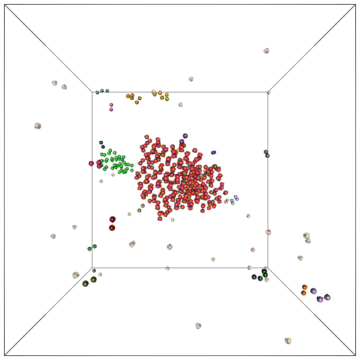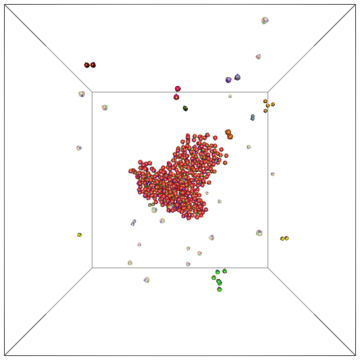
Example of a self-assembly simulation of off-lattice 'patchy-particle' DNA bricks forming a target 'top hat' structure.
However, it is well known experimentally that rather different protocols must be used for assembling DNA bricks, which entail many short DNA molecules with carefully chosen complementarity, and DNA origami, where a much longer scaffold strand is folded by shorter staple strands into the target structure. We have also designed models to probe the nucleation behaviour of DNA origamis and have shown how nucleation barriers can be tuned and even removed altogether in these systems by a judicious design of the target structure.
There is a significant role that molecular simulation can play in the process of gaining a fuller understanding of the physical properties of DNA and its self-assembly at a microscopic level, and in turn of the field of DNA nanotechnology and functional nanomaterials more broadly. DNA bricks and DNA origami both provide fascinating systems to study with many unusual features that are yet to be fully understood. In many respects, however, their properties are generalisable and, once we gain a fuller understanding of what it is that enables them to function, can be used to inform novel strategies to control pathways for self-assembly in a wide variety of systems.