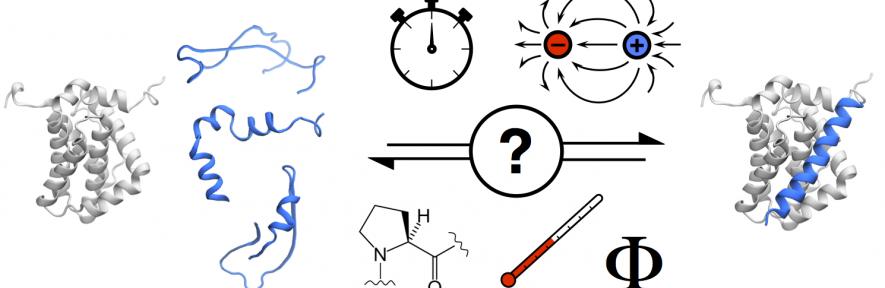
What are intrinsically disordered proteins?
Intrinsically disordered proteins (IDPs) are characterised by the lack of a singly defined three-dimensional structure. Compared to their folded counterparts, IDPs sample an ensemble of conformations. From an energy landscape perspective, this translates to a flatter, rougher surface compared to the typical funnel characteristic for folded proteins. Some IDPs gain structure upon interaction with a partner protein, a phenomenon known as coupled folding and binding.
Bioinformatic identification of IDPs can be achieved due to their amino acid composition bias (e.g. overrepresentation of charged residues, underrepresentation of hydrophobic ones). This has led to the realisation that around 30 % of the proteome of eukaryotes contains regions of disorder. Disordered proteins are more prevalent in cell signalling, processes commonly perturbed in diseases (e.g. cancer).
Due to their prevalence in biology, a thorough biophysical understanding of their properties is required1.
How do we study them?
Our lab is interested in investigating and characterising coupled folding and binding reaction mechanisms of IDPs. Information about the free and bound states of IDPs is probed using equilibrium measurements (e.g. circular dichroism, isothermal titration calorimetry). To unravel the processes leading from free to bound (and folded), information about the transition states is necessary, which we gain from kinetic studies (e.g. stopped-flow)1.
Current projects in lab
We aim at understanding general principles governing IDPs coupled folding and binding reactions. To that end we study a range of model systems. Some of the current research questions are briefly outlined below.
How different are IDPs anyway? Because of their intrinsic disorder, protein-protein interactions involving IDPs may differ with respect to their folded counterparts. For example, are they going to be associate/dissociate faster, slower or similarly? We have characterised two diametrically opposite systems, one being very fast (cMyb/KIX2) and the other slow (spectrin tetramerisation domain3). Further summarising of literature data indicates that the kinetic range of protein-protein interactions involving IDPs is similar to that of folded proteins3.
Owing to their structural heterogeneity and amino acid composition bias, IDPs may respond differently to changes in conditions compared to folded proteins (e.g. temperature, ionic strength, viscosity). Such structural effects are likely to impact their interactions with their partner proteins, but how? We extensively characterised the PUMA/Mcl-1 system and showed that standard tests to determine diffusion-limited reactions were not applicable when one partner was disordered4. We currently seek to understand the role played by electrostatics in coupled folding and binding reaction, in particular the impact of ionic strength and ions.
Mechanisms of coupled folding and binding: During a coupled folding and binding reaction, the IDP goes from unbound and disordered to bound and folded. An obvious mechanistic question is whether binding occurs first, followed by folding (so-called induced fit process) or whether folding precedes binding to the partner protein (the conformational selection paradigm). We are currently investigating this question within the pKID/KIX system.
Residual structure: Because IDPs form dynamical ensembles of interconverting structures, some subsets could in theory be more important towards binding reactions than others. Altering ensemble sampling of IDPs allows this question to be investigated. For example, we used the helix-breaker residue proline in order to assess the importance of residual helicity within the PUMA/Mcl-1 system. We showed that reduction in residual structure in the IDP PUMA largely unaffected the association rate constant, demonstrating that no particular structure was necessary for the binding reaction5.
Proline residues are also commonly found flanking intrinsically disordered regions that form helices upon binding. Are those conserved prolines present in order to modulate residual helicity in IDPs? We are currently investigating three systems to determine if a general mechanism explains the prevalence and conservation of this helix-breaking residue.
Φ-value analysis: Kinetics is unique in probing the nature of the transition of a reaction. Coupled with mutational studies, it has proven an invaluable tool to study the folding of proteins (cf. our other areas of research). We have already applied the framework of Φ-value analysis to coupled folding and binding reactions, allowing us to probe critical inter-protein contacts formed at the transition state of the reaction leading to the bound complex. We applied this strategy to both the spectrin tetramerisation3 domain and the PUMA/Mcl-1 system6 and are currently investigating the interaction between pKID/KIX.
Allostery: The KIX domain of CBP (CREB Binding Protein) is a transcriptional co-activator known to bind various IDPs at two binding sites. How does the interaction of one partner with KIX influence subsequent binding at distal sites in the context of IDP coupled folding and binding? It was found that the allosteric communication between sites, leading to tighter binding of the second partner, resulted mostly from changes in the dissociation rate constants7.
References
- Shammas, S. L., Crabtree, M. D., Dahal, L., Wicky, B. I. M., Clarke, J. (2016). J. Biol. Chem. (in press).
- Shammas, S. L., Travis, A. J., Clarke, J. (2013), J. Phys. Chem B. 117, 13346–56.
- Shammas, S. L., Rogers, J. M., Hill, S. A., Clarke, J. (2012), Biophys. J., 103, 2203–14.
- Rogers, J. M., Steward, A., Clarke, J. (2013), J. Am. Chem. Soc., 135, 1415–1422.
- Rogers, J. M., Wong, C. T., Clarke, J. (2014). J. Am. Chem. Soc., 136, 5197–5200.
- Rogers, J. M., Oleinikovas, V., Shammas, S. L., Wong, C. T., De Sancho, D., Baker, C. M., Clarke, J. (2014), Proc. Natl. Acad. Sci. USA, 111, 15420–5.
- Shammas, S. L., Travis, A. J., Clarke, J. (2014). Proc. Natl. Acad. Sci. USA, 111, 12055–60.