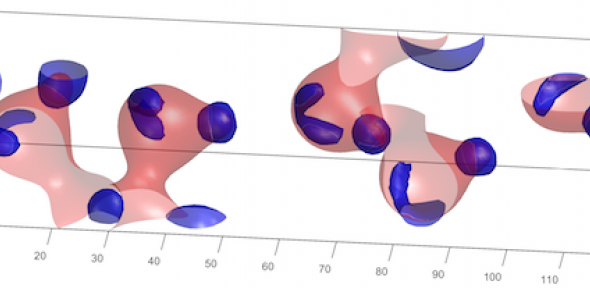
Visualisation of Hydrogen bonds and Oxide ion lone pairs in Sorbitol Molecules
Ab initio study of vibrational and structural properties of polyol glasses
With the expansion of new synthesis methods like combinatorial chemistry, the number of potential drug candidates increases yearly at a steady rate. Currently, only around 60% of discovered compounds passes the requirement of high water solubility, leaving the rest of them unattractive for further study. Techniques like grain size reduction or increase of porosity are exploited in order to enhance water solubility by increasing exposure surface area but this doesn’t affect the intrinsic solubility in water related to the high lattice energy associated with the crystalline phase observed for the majority of mentioned compounds. The amorphous state, on the other hand, shows much smaller Gibbs energy due to its highly disordered structure, making it promising for drug formation. In contrast to crystalline, amorphous state shows much greater water solubility without affecting the toxicity of the end product.
The major disadvantage of the amorphous state is its structural instability against recrystallization due to higher energy. This can lead to the loss of high water solubility, change in drug effectiveness or even toxicity. It is then clear that for any industrial application of amorphous state, stability needs to be thoroughly understood and controlled. In addition, there is no clear understanding of either the amorphous phase in general or the devitrification process (transition from amorphous to crystalline state). Experimental study utilizing techniques like time domain terahertz spectroscopy, infra-red and low-frequency Raman spectroscopy shed light on inter-molecular dynamics that control crystallization of organic amorphous solids, but the lack of atomic level investigation prevents the forming of a consistent picture.
The overall aim of this project is to study a range of polyols starting from sorbitol in its liquid, crystalline and glassy state by means of ab initio molecular dynamics simulations using Vienna Ab initio simulation package. This will allow us to describe structural properties, rotation/diffusion of molecules, vibrations, inter and intra molecular interactions, and hydrogen bonds.