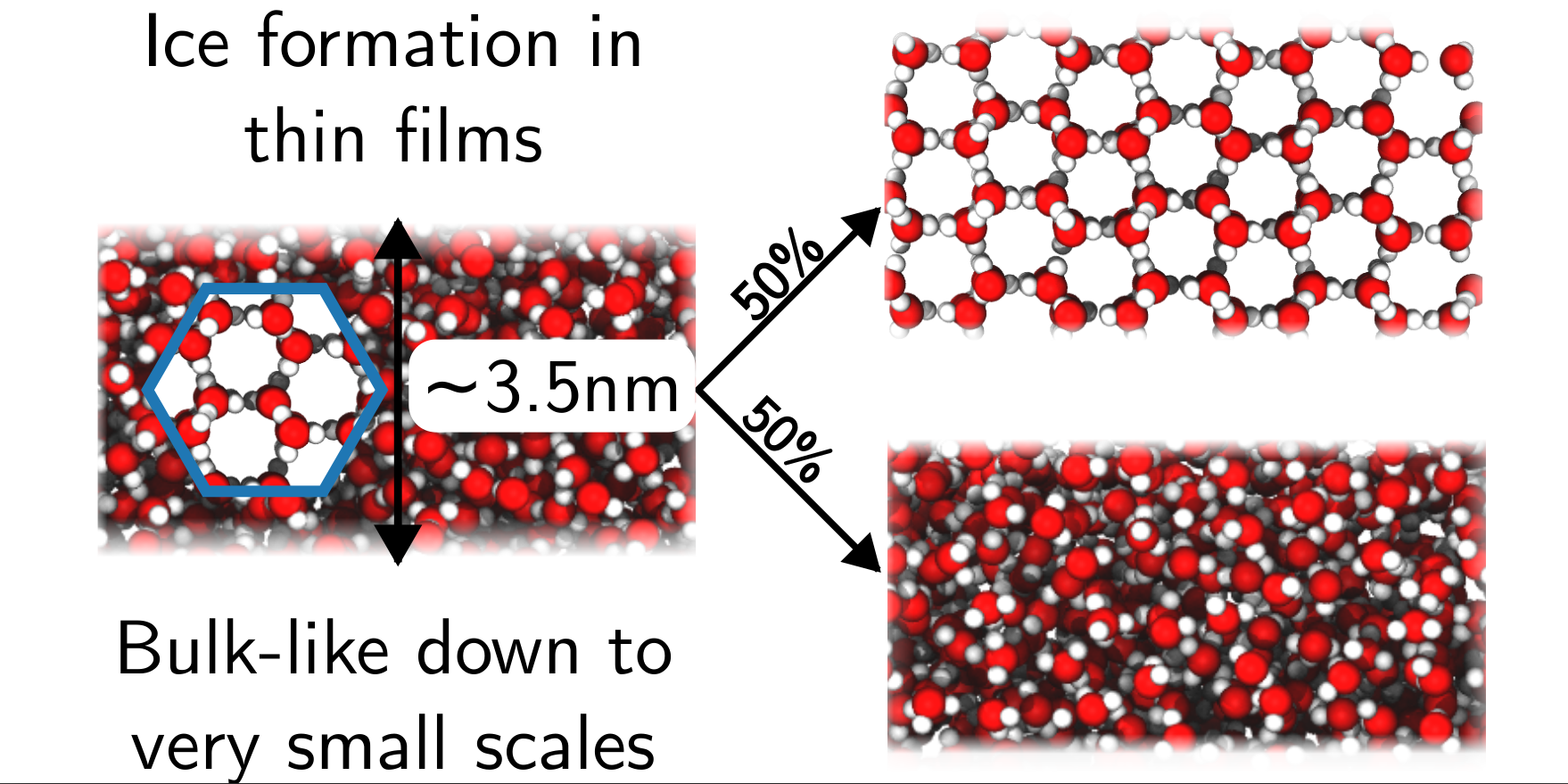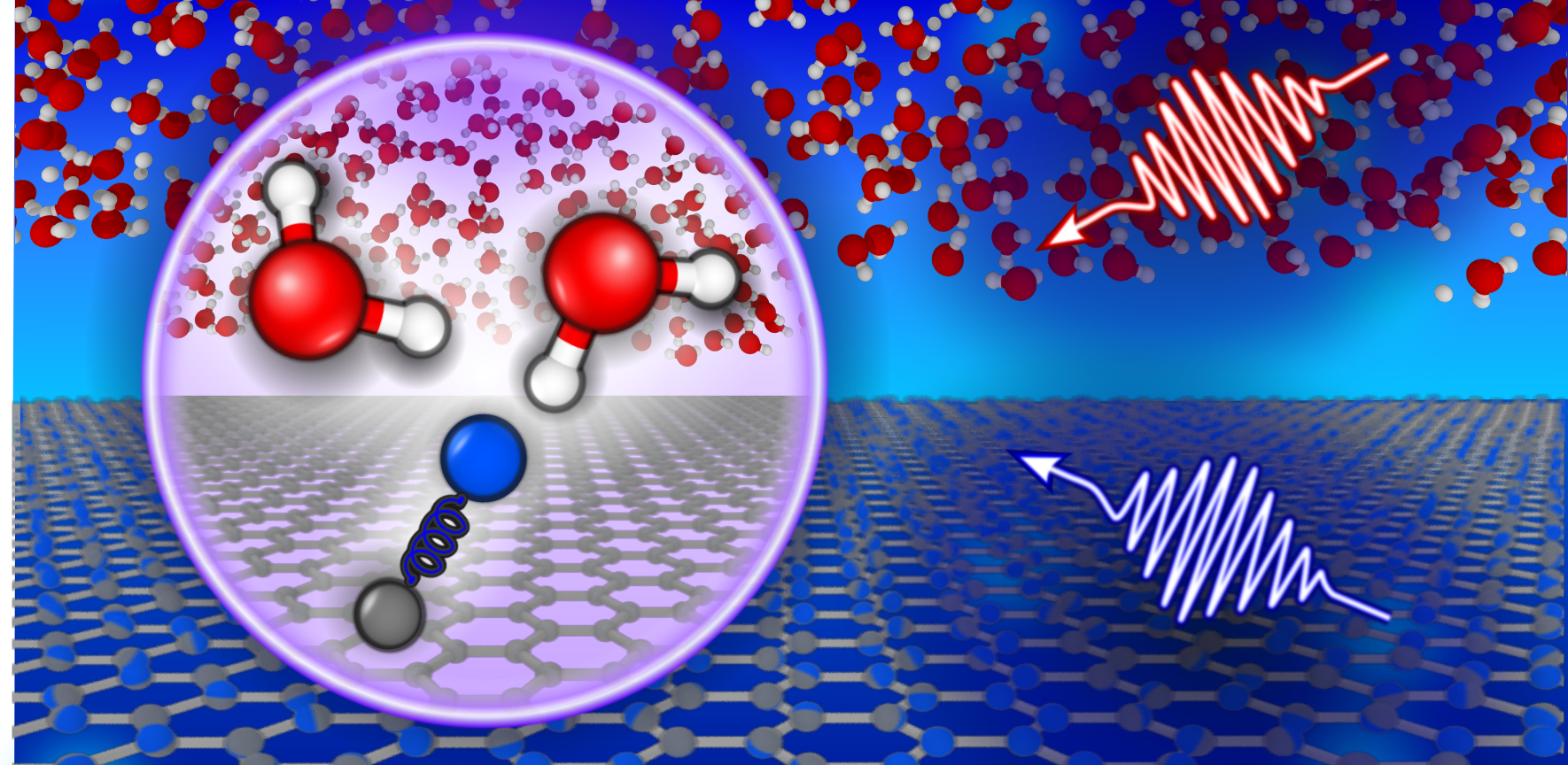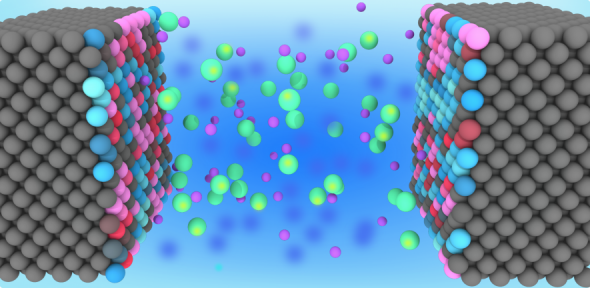
Why do we care?
As the world moves towards the decarbonisation of its electrical supply there is a significant demand for high-performance electrochemical energy storage. In order to maximise the storage capabilities of these devices a thorough theoretical understanding of the static and dynamical molecular-level behaviours that govern system efficiency is required.
Recent developments in classical molecular dynamics simulations of electrochemical cells have resulted in a renewed interest in the modelling of these systems due to a significant increase in simulation accuracy. These systems can now be modelled at a constant electric potential, compared the previous standard of constant electrode charge. This constant potential ensemble addresses the need to incorporate the trajectory of the electrolyte into the response of the electrode, allowing conducting materials to be effectively modelled.
Our work
We use simulations to deepen our understanding of the molecular-level interactions occurring at electrochemical interfaces and the pronounced impact this behaviour has on macroscopic experimental observables. Alongside both theoreticians and experimentalists, we aim to provide an improved insight into the statistical mechanics governing electrolyte behaviour at electrochemical interfaces.
Future challenges
A significant goal in this area of research is to be able to consistently produce experimental-quality analyses of measurable electronic properties through molecular simulation, to allow our computational work to seamlessly complement that which is going on in labs all around Cambridge and beyond!